Hypothesis Testing with BPP
Getting Started
Log on to the cluster and copy the data and pre-configured control files for today’s exercises:
ssh gruffalo
cd /mnt/shared/scratch/YOUR_USER_NAME/network_workshop
wget https://github.com/gtiley/RBG-Networks/raw/main/exercises/hypothesis_testing.tgz
Introduction
There are now a range of tools for detecting historical gene flow between species or populations from genomic data. This might involve searches with network models, tests with site patterns, or other lines of evidence. The goal of this exercise is to understand how full-likelihood methods can be a tool for testing multiple competing hypotheses and divergence time estimation to help us bridge the gap between patterns and processes. There are other full likelihood implementations of multispecies network coalescent or related models, but we will focus on the BPP software.
Our test data is a small sample of baobabs (Adansonia). The data, likes our previous examples, comes from Karimi et al. (2020)1.
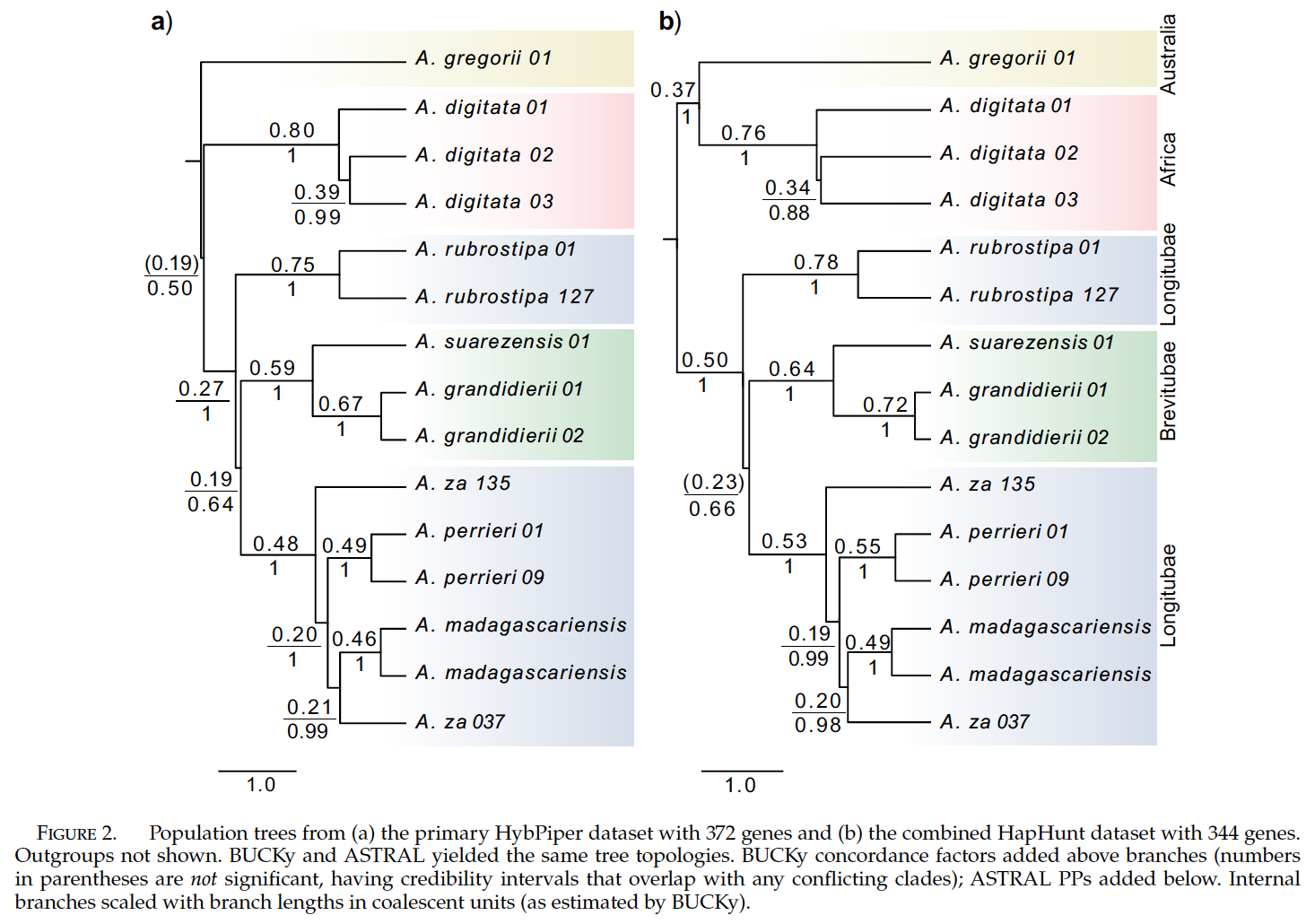
Fig. 2 of Karimi et al. 20201- Relationships among Adansonia species. Malagasy species are described in two sections based on floral morphology, section Longitubae and section Brevitubae. Notice that section Longitubae is not monophyletic. There could be many reasons for this, perhaps introgression of the genes underlying the traits. Note the strong support all nodes in the species tree based on ASTRAL’s local posterior probabilities, but many genes conflict with the backbone of this species tree.
Full-likelihood methods provide us a powerful tool because we can evaluate multiple competing hypotheses in a statistically sound probabilistic framework. However, it can be tempting to compare as many competing models as possible. In practice, this will be difficult due to computational burdens or our inability to explain the rational for every possible model. A successful application of BPP or similar full-likelihood programs relies on your knowledge of the system and their natural history, and perhaps prior analyses with a range of phylogenetic or population genetic tools. Our network hypothesis here is bolstered by a long history of chromosome investigations and previous phylogenetic results with a few Sanger loci.
How to specify a network
Formatting networks correctly can be difficult, especially when we want to specify the direction of gene flow. Luckily, BPP comes with a simple program to help create network models based on simpler binary trees. Consider our backbone topology - we will add some node labels for our convenience too. We can create a simple text file msci-create.in that provides the backbone topology as well as the branch that gene flow goes from and the branch that gene flow goes into. Multiple gene flow events can be specified here and the branch lengths of the introgression edges can be constrained if appropriate:
tree (Smic,(Agre,(Adig,(Arub,(Agra,Amad)e)d)c)b)a;
hybridization c Adig, e Agra as w x tau=no, yes phi=0.2
Here, we have allowed the divergence time of the introgression edges from their parental lineages to be different from the age of introgression. We specify an initial introgression probability of 0.2. The introgression probability will be sampled by the MCMC algorithm, so this is only a starting value. We can generate the network used for our analyses with the following command:
bpp --msci-create msci-create.in > msci-create.out
Below is the resulting network that we can add to our control file. Bonus Activity! Try to draw the network yourself from the text string and convince yourself this is correct.
(Smic, (Agre,((x[&phi=0.200000,tau-parent=no],Adig)w,(Arub,((Agra)x[&phi=0.800000,tau-parent=yes],Amad)e)d)c)b)a;
The BPP control file
All aspects of data input, results output, and the model specification are done within the control file. Here is an example for model 2 that includes episodic gene flow from A. digitata into A. grandidierii.
seed = -1
seqfile = ../../../baobab_data/baobab.100.bppDat
Imapfile = ../../../baobab_data/baobab.6taxa.spMap
outfile = model2.out
mcmcfile = mcmc.txt
speciesdelimitation = 0
speciestree = 0
speciesmodelprior = 1 * 0: uniform LH; 1:uniform rooted trees; 2: uniformSLH; 3: uniformSRooted
species&tree = 6 Adig Agra Agre Amad Arub Smic
3 2 1 2 2 1
(Smic, (Agre,((x[&phi=0.200000,tau-parent=yes],Adig)w,(Arub,((Agra)x[&phi=0.800000,tau-parent=yes],Amad)e)d)c)b)a;
phase = 0 0 0 0 0 0
usedata = 1 * 0: no data (prior); 1:seq like
nloci = 100 * number of data sets in seqfile
cleandata = 0 * remove sites with ambiguity data (1:yes, 0:no)?
thetaprior = 2 0.01 e * invgamma(a, b) for theta
tauprior = 3 0.07 * invgamma(a, b) for root tau & Dirichlet(a) for other tau's
phiprior = 1 1 * beta(a, b)
locusrate = 0 * no rate variation among loci
clock = 1 * the same rate of molecular evolutation among all branches
finetune = 1: 0.02 0.02 0.02 0.02 0.02 0.02 0.02 0.02
print = 1 0 0 0 * MCMC samples, locusrate, heredityscalars, Genetrees
burnin = 20000
sampfreq = 20
nsample = 10000
threads = 4 1 1
Parameter estimation and convergence
We can run BPP using our example control file and data. Change into the hypothesis_testing folder and you will find some sequence data baobab.100.bppDat and the individual-to-species map baobab.6taxa.spmap. Have a look to get a feel for the formatting.
Change into model1/convergence/1 and run BPP on the prepared control file by:
cd model1/convergence
ls
mkdir 1
cd 1
ls
bpp --no-pin --cfile model1.ctl
The MCMC analysis will start. It should take about 5 minutes for our simplest model with 6 species and 100 loci. The MCMC settings are too short, but necessary for the time constraints. While that is running, what information can you get from BPP’s screen output. Try completing 4 runs for a model and analyzing convergence. A helpful tool is Tracer, but BPP provides some helpful summary statistics too. An R script is provided to help plot posteriors for all parameters too. If we are satisfied, we can use one MCMC analysis from BPP or combine them to report parameter estimates. The plotting script can be executed from the command line with
cd ../../
R CMD BATCH plotPosteriors.R
Alternatively, you can run the R script from Rstudio or the R shell. A quick check of all of the parameter posteriors is that they look pretty similar. I think a good indication of convergence is a comparison of the median node heights (divergence times). If everything went well, our estimates should be very close to a one-to-one relationship.
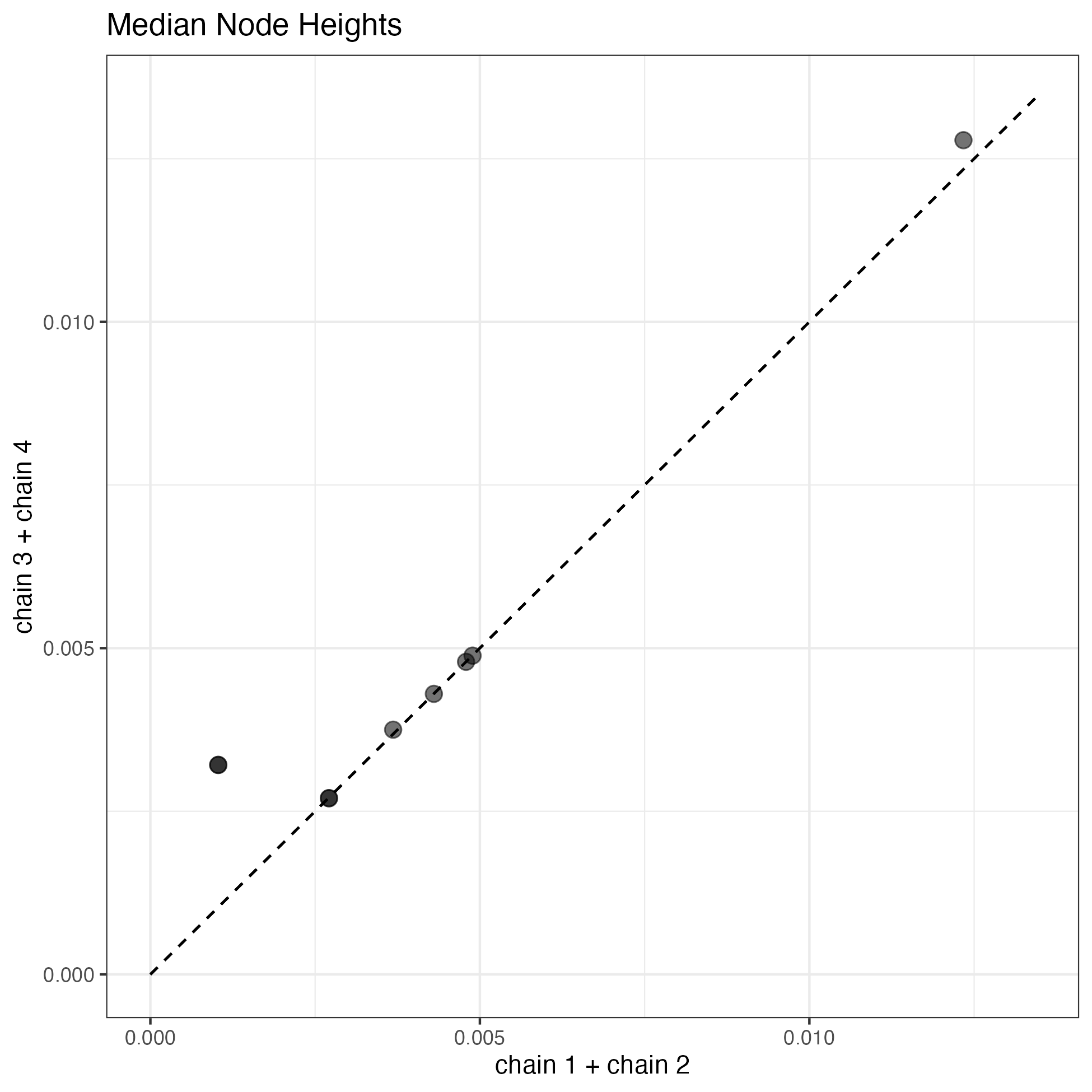
Median node heights from four MCMC runs for the two-rate MSCi model with a total chain length 50,000- The posteriors from runs 1 and 2 or 3 and 4 are combined to display in the scatter plot. The dotted line is the one-to-one line. The deviations show we should benefit from longer MCMC anlayses.
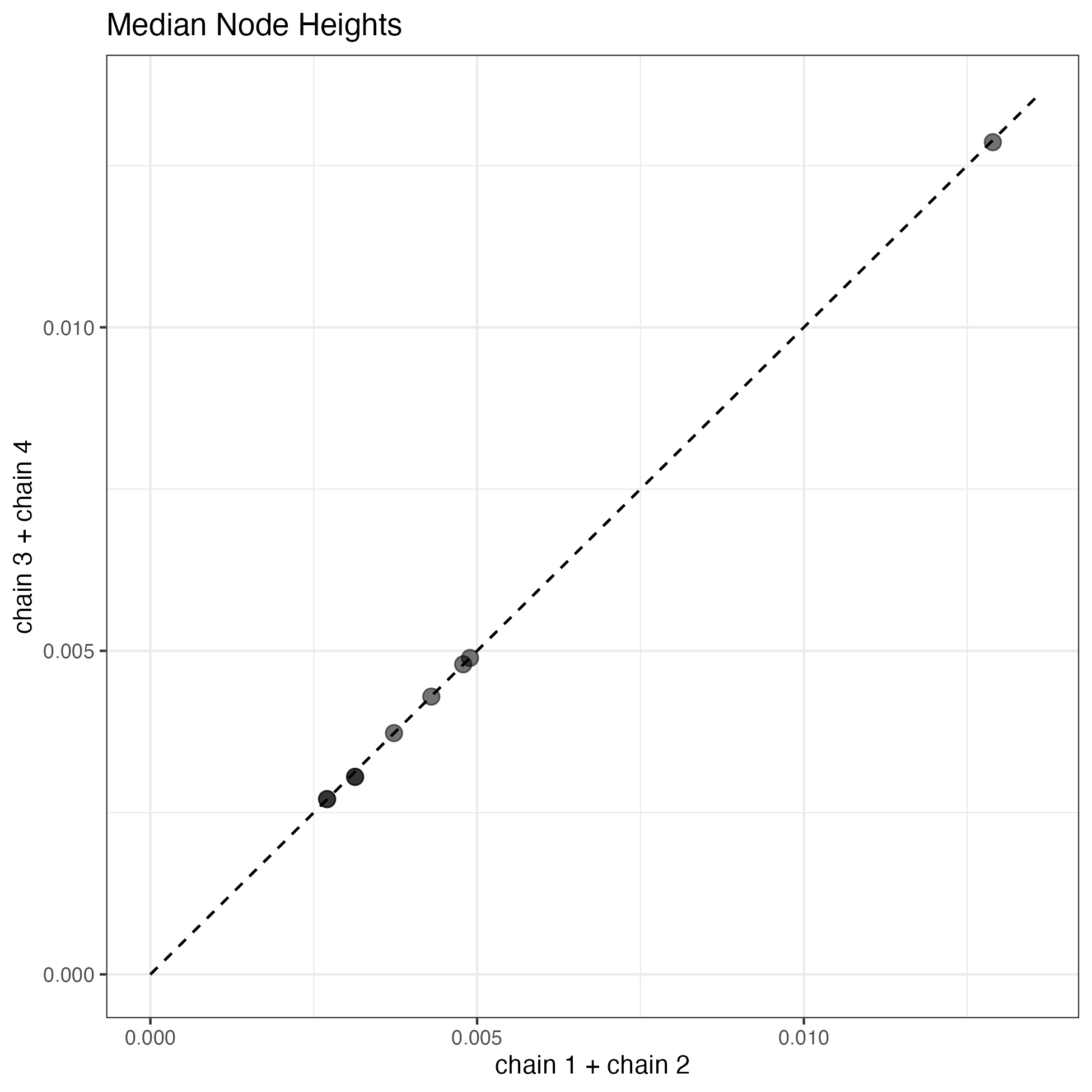
Median node heights from four MCMC runs for the two-rate MSCi model with a total chain length of 200,000- The posteriors from runs 1 and 2 or 3 and 4 are combined to display in the scatter plot. The dotted line is the one-to-one line. The one-to-one correspondance suggests a decent quality MCMC sample.
Everything should look good enough to move forward with the excercise. We could benefit from a larger sample frequency and probably an increased burnin here, but it is acceptable. For reporting our results, there are two choices:
- Report the results of the first MCMC analysis, since everything converged
- Combine the MCMC sample across runs, if we need to increase our sample size
You are fine doing either but depending on your reviewer, they might tell you to do the other. If you want to generate the BPP output for the combined sample, we can concatenate the MCMC output from the 4 runs together. Make a new file called
combined.mcmcby copying and pasting the mcmc output together, but only keep one header line. Then we need to make a new control filecombined.ctlwith some edits:
seed = -1
seqfile = ../../../baobab_data/baobab.100.bppDat
Imapfile = ../../../baobab_data/baobab.6taxa.spMap
outfile = combined.out
mcmcfile = combined.mcmc
speciesdelimitation = 0
speciestree = 0
speciesmodelprior = 1 * 0: uniform LH; 1:uniform rooted trees; 2: uniformSLH; 3: uniformSRooted
species&tree = 6 Adig Agra Agre Amad Arub Smic
3 2 1 2 2 1
(Smic, (Agre,((x[&phi=0.200000,tau-parent=no],Adig)w,(Arub,((Agra)x[&phi=0.800000,tau-parent=yes],Amad)e)d)c)b)a;
phase = 0 0 0 0 0 0
usedata = 1 * 0: no data (prior); 1:seq like
nloci = 100 * number of data sets in seqfile
cleandata = 0 * remove sites with ambiguity data (1:yes, 0:no)?
thetaprior = 2 0.01 e * invgamma(a, b) for theta
tauprior = 3 0.07 * invgamma(a, b) for root tau & Dirichlet(a) for other tau's
phiprior = 1 1 * beta(a, b)
locusrate = 0 * no rate variation among loci
clock = 1 * the same rate of molecular evolutation among all branches
finetune = 1: 0.02 0.02 0.02 0.02 0.02 0.02 0.02 0.02
print = -1 0 0 0 * MCMC samples, locusrate, heredityscalars, Genetrees
burnin = 20000
sampfreq = 20
nsample = 10000
* threads = 4 1 1
Notice the the mcmcfile is now our combined sample as input. We then change print to -1 and this will instruct BPP to bypass the MCMC and summarize the mcmcfile as the new outfile. If you every want to comment out a line in the control file, you can uses a “*”, like I did for the threads option.
Can you find the mean age of introgression for the reticulation edges and their 95% highest posterior density (HPD) intervals in the output file? What about the mean introgression probabilities? You can add the results of your analyses to a google sheet.
What if we do not have a model that we trust, but have multiple competing hypotheses? The next steps will show us techniques for comparing models, but I recommend some analysis like above across some or all of your hypotheses to check that your MCMC settings are sufficient. What can happen is that as model complexity increases (i.e. more introgression edges), you will need longer sample intervals and longer burn-ins. Settings that work well for a binary tree might be problematic for networks and this should be checked in advance before you realize the problem after computationally intense analyses.
Marginal likelihoods and model probabilities
Likelihood methods are great because they often allow us to compare and rank models. But a complication of Bayesian methods is that we have a sample of likelihoods rather than a single maximum likelihood estimate. Additionally, Bayesian estimators will be influenced by the prior distributions we imposed. There has been rigorous investigation into methods for estimating the marginal likehood where those confounding effects of the prior are factored out. The stepping-stone method Xie et al. 20112 has had good performance in a range of other phylogenetic applications and we will use it again here. BPP can generate samples of the power posteriors (Rannala and Yang 20173) that we can use to calculate the marginal likelihoods and model probabilities either from the bppr R package or by hand. We will compare 7 models today:
| Model | Description |
|---|---|
| 1 | The backbone species tree from Karimi et al. (2020) |
| 2 | Episodic introgression from A. digitata into A. grandidierii |
| 3 | Episodic introgression from A. rubrostipa into A. madagascariensis |
| 4 | Two episodic introgression events |
| 5 | Tomas’ species tree |
Marginal likelihoods are time-intensive, because they require us to generate an MCMC sample for multiple values of beta. BPP provides some tools to help you determine appropriate values of beta, but we will focus on using the bppr R package.
To estimate the marginal likelihood we need to generate the series of power posteriors. A reasonable number of steps can be hard to determine, but you can repeat the analysis for an increasing number of steps to check stability of the marginal lnL estimate. We will use 8 here to speed up the process but look at some results with 32 steps to get a feel for the effects of the number of steps on marginal lnL estimates and model selection results.
Some files have been prepared for you. Everyone will be responsible for at least one marginal likelihood calculation for a different hypothesis and model setting choice. To set up the analyses correctly, we will download the excercise files to your local computer and generate the necessary control files and folder structure and a file of the beta values called beta.txt.
setwd("YOUR_PATH/network_workshop/hypothesis_testing/YOUR_MODEL/bayes_factors")
library(bppr)
powerpb <- make.beta(8,method="gauss-quad",a=5)
make.bfctlf(powerpb, ctlf="YOUR_MODEL.ctl", betaf="beta.txt")
Notice thet some new folders have been created for you. The control files within them are identical to the starting one aside from a line at the bottom specifying a beta value for the posterior.
You will need to move the files back to the cluster at this point. Allocate an interactive node and launch the 8 jobs sequentially using the script loopFiles.sh.
—-BREAK—-
Once all of the runs are done for the 8 steps. You can calculate the marginal likelihood. Download the results (i.e. the entire hypothesis\_testing directory) to your local computer. In R, go to the bayes\_factors directory with the 8 folders for your model. For example model 1:
setwd("YOUR_PATH/network_workshop/hypothesis_testing/model1/bayes_factors")
model1 <- gauss.quad(mcmcf="mcmc.txt",betaf="beta.txt")
This works because the mcmc output from each run is called mcmc.txt. Have a look at the object created called model1 or whatever you named it. Can you find the marginal lnL and the standard error? Add those to the google sheet.
We can do the model probability calculations by hand, but we can generate the model probabilities and their 95% confidence intervals based on bootstrapping the posteriors from bppr. To get the experience, try calculating the marginal likelihoods and their probabilities on some pre-completed runs in hypothesis\_testing\_completed. For example:
setwd("YOUR_PATH/network_workshop/hypothesis_testing/completed_runs/model1/bayes_factors")
model1 <- gauss.quad(mcmcf="mcmc.txt",betaf="beta.txt")
setwd("../../model2/bayes_factors")
model2 <- gauss.quad(mcmcf="mcmc.txt",betaf="../beta.txt")
setwd("../../model3/bayes_factors/")
model3 <- gauss.quad(mcmcf="mcmc.txt",betaf="../beta.txt")
setwd("../../model4/bayes_factors/")
model4 <- gauss.quad(mcmcf="mcmc.txt",betaf="../beta.txt")
setwd("../../model5/bayes_factors/")
model5 <- gauss.quad(mcmcf="mcmc.txt",betaf="../beta.txt")
bayes.factors(model1,model2,model3,model4,model5)
Which model is best? Do you feel comfortable rejecting the putatively wrong model2 or the two-rate model4? You can see results from more steps and longer MCMC in the google sheet. Any observations about prior choice, number of loci, or differences among analyses?
Bayes factor approximation with the Savage-Dickey density ratio
Despite the rigor and computation of Bayes factors, there is some concern for preferring unnecessarily more complex models, or perhaps your error yields confidence intervals on the model probabilities that leaves the interpretation ambiguous (Tiley et al. 20234). Another tool is available to us that requires a fraction of the computing - an approximation of the Bayes factor for nested models using the Savage-Dickey density ratio (Ji et al. 20235). We can approximate the Bayes factor of model 4 versus model 3 or model 2 because model 4 adds only one more introgression edge. In R, we
setwd("YOUR_PATH/network_workshop/hypothesis_testing/model4/convergence")
posterior <- read.table(file="1/mcmc.txt",header=TRUE,sep="\t")
# The epsilon values from Tiley et al. (2023) were 0.01 and 0.001.
cutoff <- 0.01
# The number of posterior samples where the introgression probability from *A. digitata* to *A. grandidierii* is less than cutoff
nlowphi <- 0
# We are looking at the rate of episodic gene flow from node w into node x. The BPP mcmc.txt file has phi_x<-w in the header, but R does not allow mathematical symbols in headers and "<-" is converted to ".."
for (i in 1:length(posterior$phi_x..w))
{
if (posterior$phi_x..w[i] < cutoff)
{
nlowphi <- nlowphi + 1
}
}
# The posterior probability that the introgression probability of interest is less than our negligible region
posterior.probability <- nlowphi/length(posterior$phi_x..w)
# The Savage-Dickey Ratio approximation of the Bayes factor for model4/model3
sd.bayes.factor <- cutoff/posterior.probability
We can consult the tables of Kass and Raftery (1995)6 to interpret our Bayes factor result. Note this is not a log Bayes factor. I calculated about 0.02321263, which means we do not have strong support to reject the two-rate model. Try repeating the calculation with a smaller cutoff of 0.001 to ensure stability of our results. Thus, the Savage-Dickey ratio approximation can be an good strategy if your models are nested and you can generate good posteriors of the phi values. Is this stable across independent MCMC analyses of model 4 too?
As a side note, I calculated 0.02696508 when increasing the total MCMC length from 50,000 to 200,000 generations.
Limits to Biological Interpretations
In our baobab test data, we are lucky such that the patterns are corroborated by independent analyses and some expert knowledge of the system. Unknown systems do not have this advantage and the lifetime of an investigation may not be long enough to move from hypotheses to functional genomics. It can be tempting to use network methods to infer the process of hybridization or polyploidization, but I strongly caution against this. Processes other than hybrid speciation can explain high introgression probabilities (Tiley et al. 20234) and inferring extant parental species can be dubious if there are sampling gaps among species, much like how including the missing taxa might change interpretations of the analyses here. But we have seen in this limited example that full-likelihood methods perform well at characterizing the relationship of baobab species even if the model is probably much simpler (i.e. a single episodic origin) than the truth. The parameter estimates might be interesting and we can calculate probabilities for competing models. Luckily, our approaches do not seem to only favor complexity. If the model probabilities are too close for comfort and if your hypotheses are well constrained, approximation of the Bayes factor with the Savage-Dickey density ratio is simple and might be helpful for us to sort out technical artifacts of other analyses versus biologically meaningful information.
References
-
Karimi N, Grover CE, Gallagher JP, Wendel JF, Ané C, Baum DA. 2020. Reticulate evolution helps explain apparent homoplasy in floral biology and pollination in baobabs (Adansonia; Bombacoideae; Malvaceae). Syst Biol. 69:462-478.
↩ ↩2 -
Xie W, Lewis PO, Fan Y, Kuo L, Chen M-H. 2011. Improving marginal likelihood estimation for Bayesian phylogenetic model selection. Syst Biol 60:150-160
↩ -
Rannala B, Yang Z. 2017. Efficient Bayesian species tree inference under the multispecies coalescent. Syst Biol 66:823-842
↩ -
Tiley GP, Flouri T, Jiao X, Poelstra JW, Xu B, Zhu T, Rannala B, Yoder AD, Yang Z. 2023. Estimation of species divergence times in presence of cross-species gene flow. Syst Biol doi: https://doi.org/10.1093/sysbio/syad015
↩ ↩2 -
Ji J, Jackson, D, Leaché AD, Yang Z. 2023. Power of Bayesian and Heuristic Tests to Detect Cross-Species Introgression with Reference to Gene Flow in the Tamias quadrivittatus Group of North American Chipmunks. Syst Biol 72:446-465
↩ -
Kass RE, Raftery AE. 1995. [Bayes factors.] Journal of the American Statistical Association 90:773-795. - Access through JSTOR or some of the free pdfs floating about online.
↩