Variant Calling and Filtering
Genotyping can generally be taken as determining the genotype of a set of individuals, but this is conventionally used to mean reference-based alignment and variant calling of populations. We previously focused on strategies for obtaining the best-possible reference assembly. Using the same sequencing strategies across many individuals for multiple populations is not an efficient use of resources - rather we will use a single very standard Illumina library per individual. Each individual is aligned to the reference genome, which introduces error manifested as mapping uncertainty. Because a site is sequenced many times with increased coverage, piles of reads can be used to diagnose true variants versus stochastic sequencing error or other artifacts such as paralogy. This is accomplished by first identifying as many candidate variants as possible, and then using filtering strategies to reduce as many false positives as positive.
- Perform joint genotyping with GATK
- Know simple strategies for parallelizing genotyping
- Recognize the difference between site-based and haplotype-based genotyping
- Use some standard filtering tools
- Generate summary statistics from a VCF
Again, we will work in teams of two. Genotyping will use many cpus and disk space, so you will run analyses in your /work directory.
| Team (Spotter/Driver) | Mapping Score | Variant Filters |
|---|---|---|
| Mantis (Blake/Tristan F-B) | none | Basic |
| Ctenophore (Ian/Carlos) | none | Basic + depth |
| Raptor (Elissa/Tristan F) | none | Basic + depth + mac |
| Big Bluestem (Melodie) | q30 | Basic + depth |
| Polyploid Admixed Yeast (Elise/Gabi) | q30 | Basic + depth + mac |
| Scaly Tree Fern (Hannah/Karn/Marta) | q30 | Basic + depth + mac + miss |
| Mouse Lemur (Lotus/Shannon) | q30 | Brutal (GQ > 60 + no missing) |
Setup
There will be a lot of new tools introduced very quickly here.
- BWA
- Samtools/Bamtools
- Picard
- GATK
- bcftools
- vcftools
A lot of these have been pre-configured on the cluster for our use, but be warned, getting some installed correctly on your own cluster or local machine can be time-consuming and frustrating. There are some tricks in some cases, such as using a docker container for GATK. We will make extensive use of the module system today, which should handle most of our library linking problems.
Our data today and for the next few weeks will be sparrows1. There is a reference genome and 10 individuals per population. The study is focused on three populations, which we will explore when we read the paper and perform some downstream analyses. Start by copying today’s folder with the data and some pre-configured scripts over to work.
cd /work/YOUR_ID/evolutionaryGenomics
cp -r /hpc/group/bio790s-/evoltuoinaryGenomics/populationGenomics1 .
cd populationGenomics1
ls
You should see the following folders
- reference - the reference genome sequence and some metadata tables are in here
- bams - we will generate the alignments here
- genotyping - where the genotyping will happen
- filteredVCF - a place to keep the final multi-sample vcf files
Reference Alignment
BWA was used previously be Juicer to align the Hi-C reads to the contigs. Now, we will think a bit more of what is going on here. The reference genome is indexed using a suffix array which is then searched against by your millions of short reads compressed by an algorithm called Burrows-Wheeler Transform. It is a well-characterized way to reduce complexity in string search space used also by Bowtie. Perhaps BWA became so popular because it was from the same group also thinking about the downstream processing tools Samtools and ultimately how we represent genomic data today with all of the quality score goodness. In most cases there is no need to change the defaults with BWA, let it do its thing and we can think about errors in post. Keep in mind that for each alignment, BWA produces a mapping quality score (mapq). Like other quality scores we have seen, these are Phred-Scaled probabilities that an alignment is incorrect. I have often seen mapq of 30 treated as sufficient, which is saying that the probability of an error of 0.001 is acceptable.
Read groups are added when aligning with BWA too. Read groups provide metadata, such as the individual sequenced and the library. If you have multiple libraries, you need read groups so PCR duplicates can be correctly identified and omitting them will cause problems. This is annoying to fix later on, so I always recommend adding read groups, even if you have a single library per individual.
BWA can also be chained together with Samtools. This is largely to reduce the need for intermediary files by piping output from one program to another. Notice we will bypass the sam format altogether and jump immediately to the binary bam format. The bam will also be sorted, which is necessary for reading by downstream tools like GATK. The command will look something like this for a single individual.
bwa index reference.fasta
bwa mem -R '@RG\tID:id1\tSM:sp1\tPL:illumina\tLB:lib1' -t 4 reference.fasta ind1_R1.fq.gz ind1_R2.fq.gz | samtools view -bS -F 4 - | samtools sort - -o ind1.sorted.bam
But we have to do the alignment step for 30 individuals! So, our strategy will be to do all of the indexing first, including for downstream samtools applications and for GATK (using Picard). Then, we will use a script to launch all of the BWA jobs for us. This builds off of the loopFiles examples from our first class. I provide a Perl version here, but can you edit the Python loopFiles template to achieve the same goal? Normally you would do these steps, but since it is repetitive with past exercises, I have made the bams in advance - you can skip the next code block.
cd reference
emacs indexGenome.sh
sbatch indexGenome.sh
cd ../bams
perl bwa.pl
Genotyping
Haplotype-based genotypers are arguably regarded as the best based on benchmarking studies and include GATK and FreeBayes. They also have to make some assumptions that may not always be appropriate though. bcftools is also capable doing the genotyping for you and will be a lot faster. It can similarly solve haplotype phase, but does not perform this haplotype-based realignment of active regions, it calculated the genotype likelihoods per-site from the bam directly. ANGSD is a very intersting option that uses uncertainty in the data by weighting with genotype likelihoods rather than trying to make calls - this can be advantageous in some cases but not always compatible with downstream analyses. We will use GATK in-part because of it’s popularity but remain open that other choices are good and perhaps preferable under some scenarios.
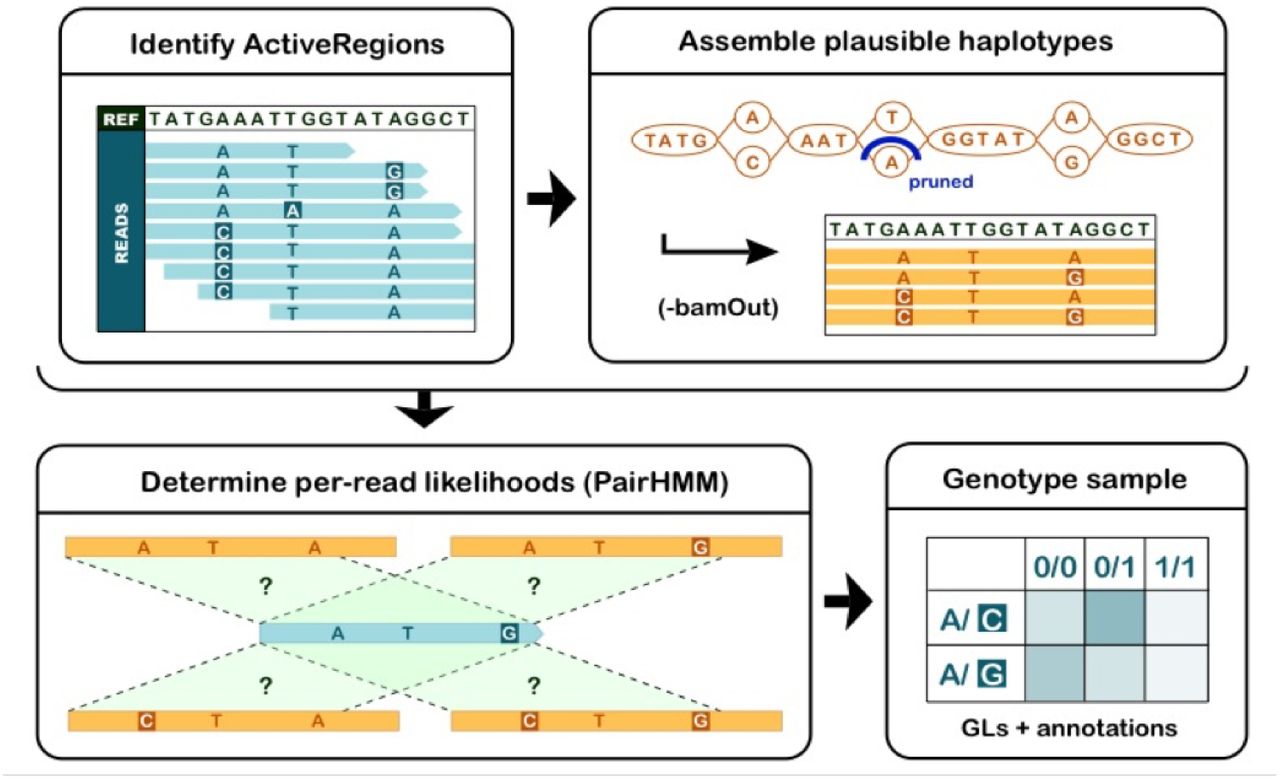
Fig. 2 from Poplin et al. 20182- Local re-alignments are performed in regions determined to have variation from the bam (active regions). The read distributions are used to detect haplotype combinations and ultimately estimate the genotype likelihoods.
The basic commands look something like this:
picard MarkDuplicates I=ind1.sorted.bam O=ind1.marked.bam M=ind1.metrics.txt
gatk HaplotypeCaller -R reference.fasta -I ind1.marked.bam -O ind1.g.vcf -ERC GVCF --native-pair-hmm-threads 4
But again, this step needs to be repeated for all individuals! Remember to edit your email in the Perl script before running! Have a look at how we loop over the individuals based on the available bams and write out the relevant SLURM scripts that will do the work for us. It is possible to accomplish this with job arrays through SLURM too, but this is simply how I learned to do things. If you are using the reads already filtered for mapping quality, you will need to change the script accordingly. I also commented out the final sbatch command the submit the jobs in case you want to run it and see what happens first. When you are ready to submit all of the jobs, uncomment this.
cd ../genotying
perl gatkGVCF.pl
The output will be that each individual gets a .*g.vcf file. These are intermediary formats with information about the variants discovered within individuals that will be pooled with the other samples in the joint calling step. This will take some time, but when it is done, we move on to the joint calling that takes a slightly different strategy to parallelization.
gatk GenomicsDBImport --genomicsdb-workspace-path database --intervals interval --sample-name-map sample.map --tmp-dir=temp --reader-threads 4
gatk GenotypeGVCFs -R reference.fasta -V gendb://database -O interval.joint.vcf
First, all of the individual .g.vcf files are collected into a database where those individual genotype likelihoods estimates are used in a multi-sample model to call variants in the population. To speed up this process, we can define a specific interval on which to operate. The intervals would be headers in reference fasta file, hopefully chromosome names. In our case we will loop over the scaffolds that have been assigned to chromosomes and not worry about the unanchored bits of the assembly. It is my personal opinion that those unanchored bits are of limited use and often raise more issues than provide resolution to the question at hand. Those databased are then passed to the joint genotyping step - GenotypeGVCFs. These steps are implemented in the provided Perl script. The script also generates some commands for combining the interval VCFs together, filtering out everything that is not a biallelic SNP, and then filtering away things that do not pass what we are calling basic filters. These are very rough recommendations from the GATK devs, that most people deviate from in practice to fit their needs, but they are a good place to start. If you have the Brutal filter, you will need to add an extra filter expression (“GQ < 60”).
cd ../jointGenotyping
perl gatkJoint.pl
#after joint calling is finished
sbatch gatkFilter.sh
Filtering
We have already done a bit of light filtering based on some features such as strand biases and mapping, but setting some filters require probing of our candidate variants. Perhaps most important is depth. We want to get rid of sites with low mean depth and mask variants in individuals with low depth. High depth can be a problem too if it is greater than expected, as it likely represents paralogy or other mapping issues3. A number of depth filters have been proposed, but a simple rule is 2x the mean depth. This should address problems from recent or tandem duplicates - older duplicates that have moved throughout the genome should hopefully have enough difference in their mapping scores to be viable. We will also look for odd individuals that we would be suspicious about based on heterozygosity and allele balance. We will set some threshold of missing data too.
Here vcftools is used to generate a number of summary statistics about the post-GATK-filtered vcf. We can then take those files to look at the distributions in R. We will do this together in real-time, but I largely follow the recommendations from the speciation genomics workshop. These are the flags we will use to get some helpful information:
- –freq2 (allele frequency - relevant to mac filter)
- –depth (mean depth of each individual)
- –site-mean-depth (mean depth of each site)
- –site-quality (mean genotype quality score - Phred-scaled difference between best and next best genotype)
- –missing-indv (missing data per individual)
- –missing-site (missing data per site)
- –het (heterozygosity per individual)
The relevant output files can be generated with vcfStats.sh in the vcfStats folder. The filters will then be applied in filterVCF.sh. You should remove or add filters as necessary to match your teams filtering strategy. The Brutal filters should set MISS=1. bcftools are also used here to count the number of variants before any filtering and how many are left after filters. How do your results look? We also call bcftools three times to generate vcfs with subsets of individuals - these will be used in three weeks and getting to this step will take some time.
Caveat
Note that we have done this in a way where GATK ignores parts of the genome where it does not find variants. This can be problematic if you need to know something about callability, and even for some popgen stats. If you need to differentiate invariant sites from a lack of information about a site, use -ERC BP_RESOLUTION in the HaployeCaller step. Be warned this will generate much larger vcf files, since it will be every site in the reference genome.
References
-
Elgvin TO, et al. 2017. The genomic mosaicism of hybrid speciation. Sci Adv. 3:e1602996. ↩
-
Poplin R, et al. 2018. Scaling accurate genetic variant discovery to tens of thousands of samples. bioRxiv doi:10.1101/201178 ↩
-
Li H. 2014. Toward better understanding of artifacts in variant calling from high-coverage samples. Bioinformatics 30:2843-2851. ↩