Divergence Times and Rates of Evolution
Species trees calibrated to absolute time are important for generally understanding speciation in the context of geological and climate change. These time-calibrate trees are also ultrametric, the root-to-tip distance for all terminal nodes is the same, and are necessary for estimating diversification rates, rates of character evolution, or rates of change of other types of data such as gene gain and loss. We will examine the approximate likelihood approach of dos Reis and Yang (2011)1, which is one of a few, but arguably the most statistically rigorous, approach to estimating divergence times with whole-genome data for many tips. It is implemented with the MCMCTREE program that is included in PAML2. Although, the general theory has been around since 19983, this approach is used in many contemporary studies because it is both fast by relying on approximations from a Taylor expansion of the likelihood function and accurate since branch lengths can be estimated with high precision in genome-scale alignments. We will use the alignment for six primate species plus an outgroup (Fig. 1) to explore the effects of model (independent versus autocorrelated rates) and calibration choice.
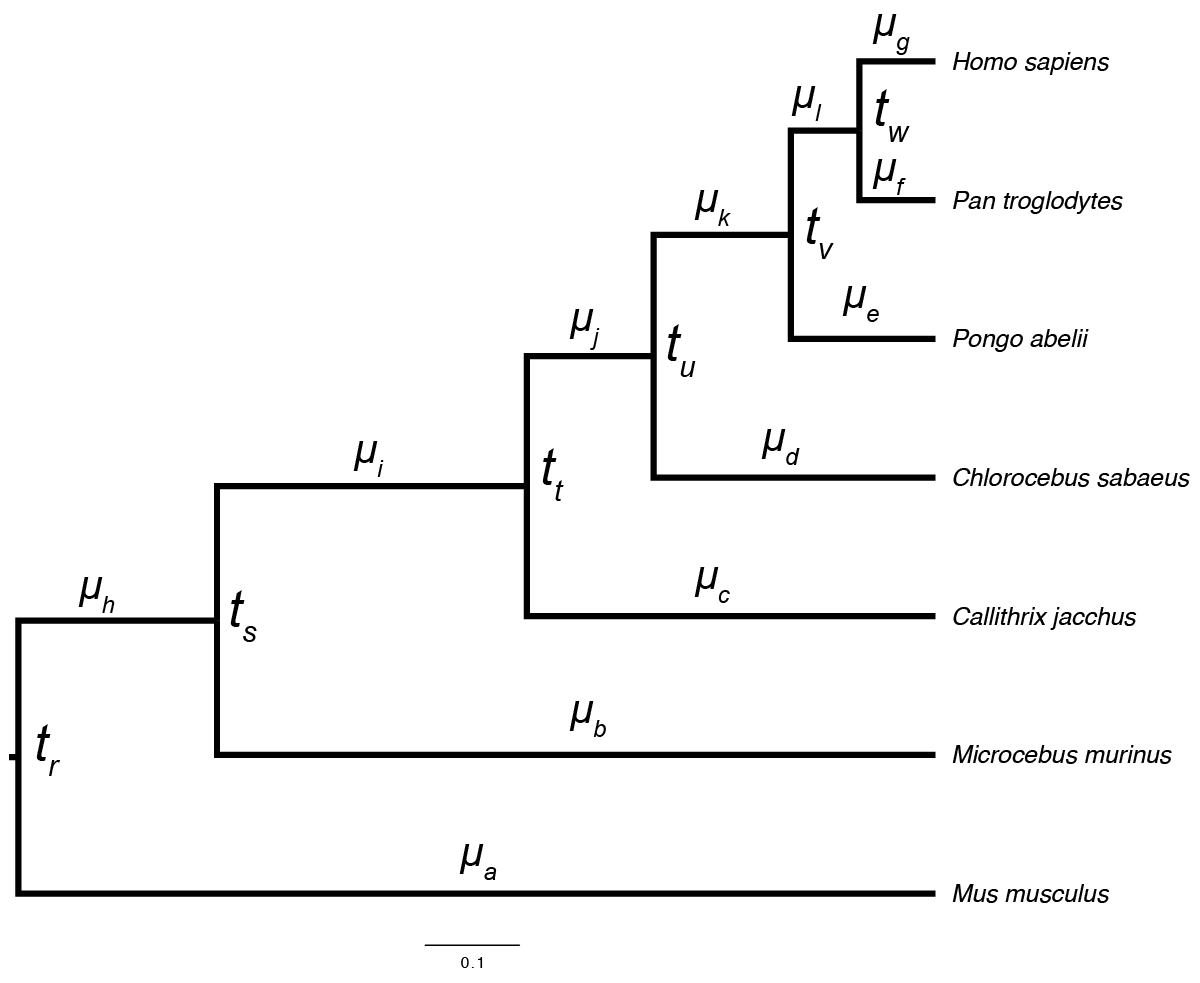
Fig. 1- Species tree for seven taxa. A relaxed-clock model will have 6 divergence times (t) and 12 rates ($\mu) to estimate.
We will continue working in teams to examine these different scenarios. Changing the model is easy as you will see. Setting up the calibrations can be tricky and requires some knowledge of the fossil record. I have prepared in advance a tree where all nodes have high-quality calibrations, but two other cases where we can see what happens when a very precise calibration is placed towards the tips or the root and the others are uncalibrated. The root node always has a fairly vague calibration not particularly grounded in primary fossil evidence, but to make the estimator behave reasonably.
| Team | Model | Calibration |
|---|---|---|
| Mantis (Blake/Melodie) | Independent (clock = 2) | All |
| Ctenophore (Carlos/Gabi) | Independent (clock = 2) | Root + Primate |
| Raptor (Elissa/Shannon) | Independent (clock = 2) | Root + human-chimp |
| Big Bluestem (Ian/Tristan F-B) | Autocorrelated (clock = 3) | All |
| Polyploid Admixed Yeast (Elise/Marta) | Autocorrelated (clock = 3) | Root + Primate |
| Scaly Tree Fern (Hannah/Tristan F) | Autocorrelated (clock = 3) | Root + human-chimp |
Getting the Gradient and Hessian
The approximate likelihood method for requires estimating the branch lengths by ML. First, you run a control file for MCMCTree that specifies to generate a second control file for BASEML, which does the ML optimization under the specified model. You would want to read the PAML manual carefully to understand all options, but here is a brief rundown of the control file mcmctree-outBV.ctl
seqfile = ../input/mn.0.phy
treefile = ../input/7tax.calibrations.all.tre
ndata = 1
seqtype = 0 * 0: nucleotides; 1:codons; 2:AAs
usedata = 3 * 0: no data (prior); 1:exact likelihood;
* 2: approximate likelihood; 3:out.BV (in.BV)
clock = 3 * 1: global clock; 2: independent rates; 3: correlated rates
model = 4 * 0:JC69, 1:K80, 2:F81, 3:F84, 4:HKY85
alpha = 0.5 * alpha for gamma rates at sites
ncatG = 5 * No. categories in discrete gamma
cleandata = 0 * remove sites with ambiguity data (1:yes, 0:no)?
ndata = 1is specifying that we have 1 partition. If you had multiple data partitions (e.g. coding 1st and 2nd positions versus 3rd positions), ndata let’s the program know how many partitions to read in. A BASEML file would be generated for each file seperately which could be run independently to parallelize the computation.seqtype = 0specifies that we use single bases as data and to use nucleotide substitution matrices. Using codon models is often not done for divergence times (the optimization and be difficult and perhaps the MLEs will not adequate for heterogenous codon data). Rather, amino acids are used at deep phylogenetic levels, like the butterflies, but using the amino acid models with MCMCTree takes a few extra steps not covered here, but are not difficult to follow once you have some intuition about the models and software.usedata = 3let’s MCMCTree know to prepare the appropriate BASEML files. When we are ready to run MCMCTree for divergence time estimation with the optimal gradient and Hessian, this number will change to 2.clock = 3is irrelevant for now because we are generating the BASEML files first. But When running MCMCTree inrunmode = 2, this will affect how rate variation among branches is modeled.model = 4 alpha = 0.5 ncatG = 5is selecting the HKY substitution model with gamma-distriibuted among-site rate variation. The gamma is discretized using 5 rate categories.
Now that some parts of the input have been explained, we will run MCMCTree, then BASEML, to get the gradient and Hessian. I have made a compiled version of PAML and its various programs available in our group folder on the cluster. Write a submission file to execute the following command from within the macroevolution2/hessian folder after you copy the macroevolution2 folder over to your work directory.
/hpc/group/bio790s-01-f21/evolutionaryGenomics/programs/paml4.9j/bin/mcmctree mcmctree-outBV.ctl
Once the job is complete, you will see three files with the prefix tmp0001. tmp0001.txt is a copy of the sequence alignment and temp00001.trees is a copy of our tree file, but with the annotations about fossil calibrations removed. The third is a new control file for BASEML tmp0001.ctl
seqfile = tmp0001.txt
treefile = tmp0001.trees
outfile = tmp0001.out
noisy = 3
model = 4
fix_alpha = 0
alpha = 0.5
ncatG = 5
Small_Diff = 0.1e-6
getSE = 2
method = 0
This is a repeat of the model setup, but with some additional optimization options for BASEML. You should never need to modify the tmp*.ctl files. Maybe something to note is fix_alpha = 0, which lets alpha be optimized as part of the likelihood function. You could fix the value of alpha by switching to 1. Obtaining the MLEs of other parameters while fixing one is useful for obtaining the marginal likelihoods over a range is helpful for teaching demonstrations, or investigating when you think the likelihood surface for a particular parameter is flat or has multiple optima. Edit your submission script to run BASEML on tmp0001.ctl
/hpc/group/bio790s-01-f21/evolutionaryGenomics/programs/paml4.9j/bin/baseml tmp0001.ctl
The optimization should not take long as there are only 7 taxa and the alignment has been reduced to 1 million sites. There will be a lot of output files, but the important one is rst2. Why are things named this way? Legacy program development - one of those things you just have to learn along the way. rst2 is our output file with the gradient and Hessian for MCMCTree. We just need to rename it and then we are done with this step.
cp rst2 in.BV
Important: We only had 1 partition to deal with here, but what if you have many and each temp*.ctl is going to make something we need called rst2? To prevent files from being overwritten, run each temp control file in a sperate directory for each partition (e.g. 0, 1, .., n). The in.BV file just becomes the concatenated input of these separate partitions cat 0/rst2 1/rst2 ... n/rst2 > in.BV.
Estimating the Posterior with MCMC
We are now ready to run MCMCTree. Move into the macroevoltuion2/posterior folder to find a new control file mcmctree.partitioned.ctl
seed = -1
seqfile = ../input/mn.0.phy
treefile = ../input/7tax.calibrations.4.tre
mcmcfile = mm.0.mcmc.txt
outfile = mm.0.out.txt
ndata = 1
seqtype = 0 * 0: nucleotides; 1:codons; 2:AAs
usedata = 2 * 0: no data (prior); 1:exact likelihood;
* 2:approximate likelihood; 3:out.BV (in.BV)
clock = 3 * 1: global clock; 2: independent rates; 3: correlated rates
RootAge = '<1.0' * safe constraint on root age, used if no fossil for root.
model = 4 * 0:JC69, 1:K80, 2:F81, 3:F84, 4:HKY85
alpha = 0.5 * alpha for gamma rates at sites
ncatG = 5 * No. categories in discrete gamma
cleandata = 0 * remove sites with ambiguity data (1:yes, 0:no)?
BDparas = 1 1 0 * birth, death, sampling
kappa_gamma = 6 2 * gamma prior for kappa
alpha_gamma = 1 1 * gamma prior for alpha
rgene_gamma = 2 40 1 * gammaDir prior for rate for genes
sigma2_gamma = 1 10 1 * gammaDir prior for sigma^2 (for clock=2 or 3)
print = 2 * 0: no mcmc sample; 1: everything except branch rates 2: everything
burnin = 100000
sampfreq = 100
nsample = 10000
finetune = 1: .1 .1 .1 .1 .1 .1 * auto (0 or 1): times, musigma2, rates, mixing, paras, FossilErr
- If you have the independent rates model assigned to your group, remember to change clock=3 to clock=2
- Remember to copy and edit the tree file for your calibration configuration and change the file name appropriatly
- New parameters such as BDparas, rgene_gamma, and sigma2_gamma, are the tree prior (speciation=extinction), rate variation among partitions, and rate variation among branches. We will forego a careful discussion of priors for now, but selecting these requires some carefull thought and synthesis of the literature to center the distributions while keeping them sufficiently vague.
- We set print=2 so we get the rates of evolution per-branch. I often care about them, but there will be many rate parameters for large phylogenies and if you see them as little more than a nuisance, you can set print=1. It is good practice though to check the sampling efficiency and convergence of rate parameters where possible though.
- burnin is the number of generations skipped before MCMC samples are retained. After the burnin, we save every 1000 samples until we have 10000. There is often little value in collecting more than 10000 samples. IF you need longer for convergence, consider increasing the sampling frequency. I have put the burnin and sampling frequency very low to speed up our exercise, but you would want to increase both to at least 1000000 and 1000 and a good starting place is to make burnin 10% of your total run length.
Also, we will get into convergence a little later, but to evaluate convergence we need multiple independent runs. Notice there are four folders (1,2,3,4). Edit the control file for your clock model and tree, then copy it into each one. You can make 4 different submission scripts and launch all 4. Don’t forget to copy the in.BV file into each folder! Each job should take less than 5 minutes.
/hpc/group/bio790s-01-f21/evolutionaryGenomics/programs/paml4.9j/bin/mcmctree mcmctree.partitioned.ctl
The jobs should get to work and you will see the MCMC files grow as the sampling progresses. While we wait, let’s think about convergence.
Convergence
Any estimator will give you an answer, but how do your know it is reliable. And if you have enough clout you often don’t have to say much about it in your manuscript text, but convergence is a low-hanging fruit that reviewers like to feint panic about. Tracer is a great tool for evaluating posteriors in the mcmc files interactively, but maybe clunky if you need to make publication-quality graphics. Try downloading your macroevolution2 folder to your local computer after the analyses have finished. Go into the convergence folder and try running the combinePosteriors.pl Perl script. That will concatenate the posteriors from run1 with run2 and run3 with run4. You can then run the R code PosteriorPlots.R, after setting the correct working directory in the script. This will give you some other thoughts on how to inspect convergence beyond Effective Sample Size (ESS), which can be a path to ruin for very large phylogenies with a lot of autocorrelation. Instead, we can look for a simple one-to-one correlation between the runs (Fig. 2) for divergence times and rates. The R script will generate these plots, although it would need some heavy modification for other trees and models.
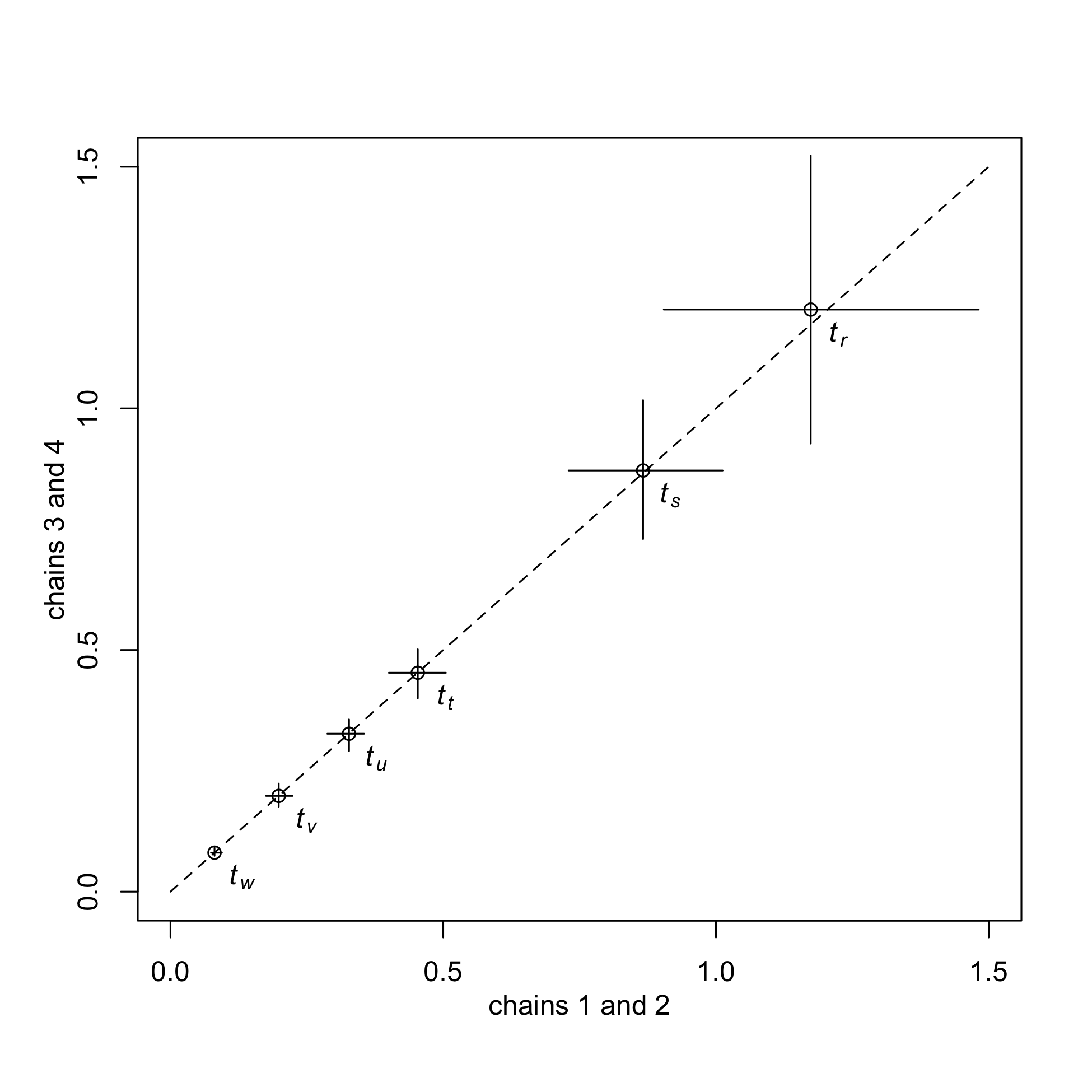
Fig. 2- Divergence Times Compared between Independent Runs for the same Model. Points are posterior medians an error bars are 95% Highest Posterior Density (HPD) intervals.
Concluding
Convergence should look ok, it would benefit from longer runs but this will be very clean data. The runs with all calibrations will converge quickly but the runs with only two will have a lot more error and be messy. For now, take the means and 95% HPDs of divergence times from run 1 (in out.txt or can be calculated from mcmc.txt in R) and enter them into the google sheet.
If we finish in time, we will plot the results at the end of class. If you need longer to finish runs, please add results when they are done and we can circulate plots later.
References
-
dos Reis M, Yang Z. 2011. Approximate likelihood calculation on a phylogeny for Bayesian estimation of divergence times. Mol Biol Evol. 28:2161-2172. ↩
-
Yang Z. 2007. PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol. 24:1586-1591. ↩
-
Thorne JL, Kishino H, Painter IS. 1998. Estimating the rate of evolution of the rate of molecular evolution. Mol Biol Evol. 15:1647-1657. ↩