Genome Architecture
Disclaimer: We are only examining, maybe what could be called macrosynteny. We use protein-coding genes as our anchor points and are concerned only about the relative order. We will gloss over some of the intermediary steps today to focus on running the different software components, but can discuss strategies for going between the analysis and visualization.
We will look at a subset of moss data from Carey et al. 20211. This data is unrelated to our readings this week, but relevant to placing the 1R and 2R events in vertebrate evolution in Nakatani et al. 20212. While they develop a probabilistic model of what the ancestral genomes look like, we will use a simple heuristic approach that can still be powerful. For the mosses, the main issue is that data suggests a single whole-genome duplication in Ceratodon purpureus and two whole-genome duplications in a very different species Physcomitrium patens. Our goal will be to use syntenic information to infer if these events were independent of each other or shared.
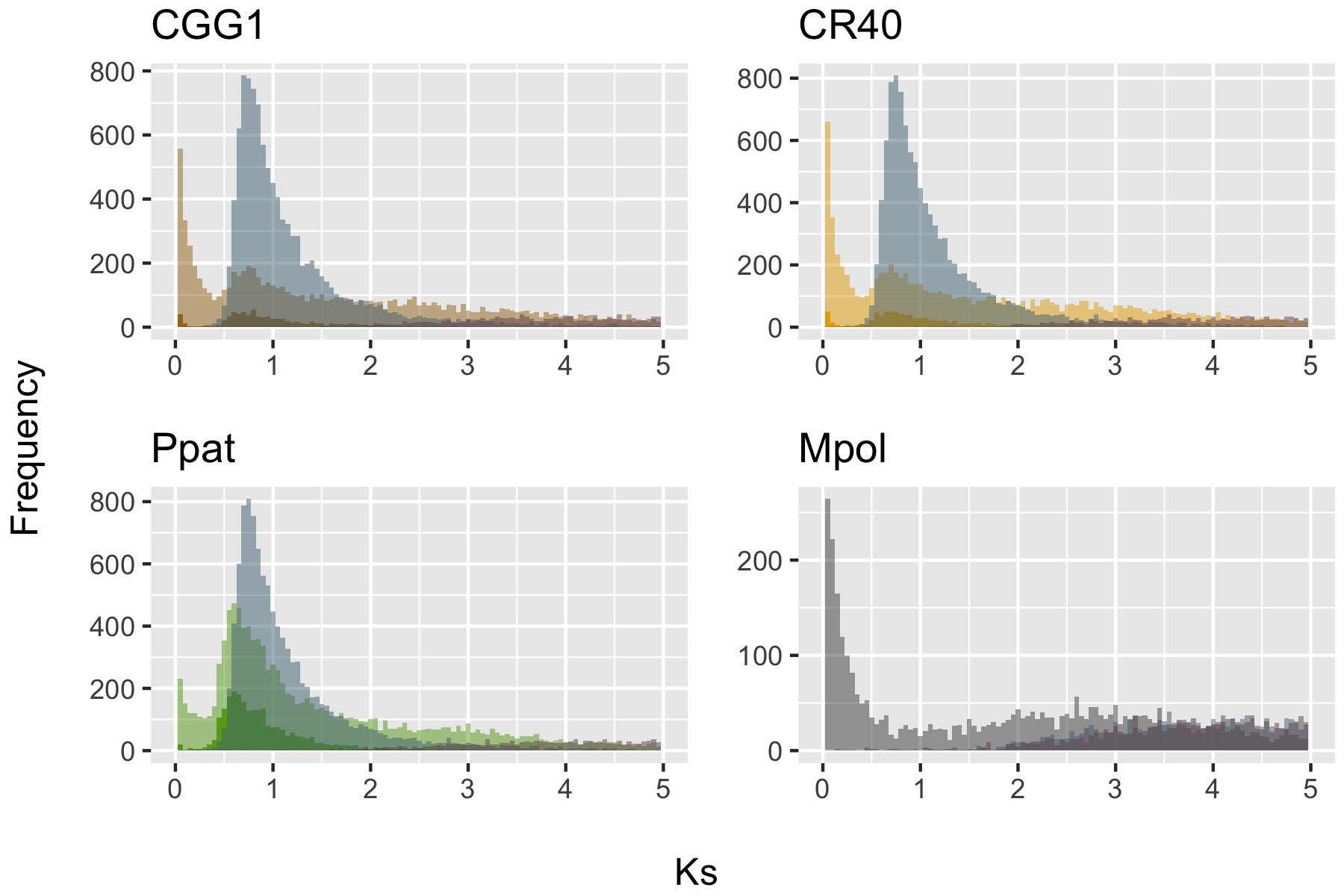
Fig. S2c from Carey et al. 20211- Peaks in the distribution of Ks indicate whole-genome duplication. Because paralog peaks (the lighter color for all paralogs and darker color for syntenic paralogs) occur more recently that the ortholog divergences (the blue-greyish color), we might assume the events were independent and not shared.
Homology
Extracting information about architecture requires us to infer homology within and between genomes. Although there are options, this typically means BLAST. The first step in examining synteny will be an all-by-all blast, where all genomes are blasted against themselves and everybody else. We do all pariwise combinations because which genome is the query or the database can affect the e-values. You will set up your own blast jobs on the cluster. Remember to copy the directory macroevolution1 to your work directory. A few hints:
- To set up the paths to BLAST and link the libraries correctly, add this module load line
module load NCBI-BLAST/2.11.0-rhel8 - Before blasting, you will need to make the databases for each genome. This step is very fast
makeblastdb -dbtype prot -in Ppat.protein.processed.fasta -out Ppat makeblastdb -dbtype prot -in CR40.protein.processed.fasta -out CR40 - We are using blastp because these are amino acid sequences. Here is one line, and you will need to do the other combinations. Be sure to correctly specify the number of threads you need in the SLURM directives.
blastp -num_threads 4 -max_target_seqs 10000 -max_hsps 1 -outfmt 6 -evalue 1e-40 -query Ppat.protein.processed.fasta -out Ppat.CR40.out -db CR40
For the sake of time, these output files are already available in the folder blast_outfiles. You will see each one is a tab-delimited table with the query and database hit. Because we specify a strict e-value cutoff in the blast options, we could consider all of these to be reasonable. These hits include, orthologs, paralogs, and errors - and alone do not tell us much. The hits can be used to discover the syntenic regions with some reasonable degree of error though.
Synteny
We will use the program MCScanX. There are other options, but this has been the tool of choice for the plant genomics community. It has now moved to github, which means you can download it to your local maschine using git clone. Once downloaded, you should be able to change directory and simply run make to compile. I suggest placing this exercise in its own directory.
cd ~/evolutionaryGenomics
mkdir Macroevolution1
cd Macroevolution1
git clone https://github.com/wyp1125/MCScanX
cd MCScanX
make
Now, download the MCScanX_Example folder from the cluster to your computer. We will change into that folder and run MCScanX a few times.
cd MCScanX_Example
../MCScanX_h moss/moss -s 10 -m 20 -b 0
You will now see some output about the number of syntenic pairs between and within genomes. Does this look like a lot or a little? A few options here, -s is the number of genes needed to seed a region. Blocks less than 10 are ignored. -m is the number of interleaving genes allowed (the Manhattan distance). This means 20 non-syntenic genes are allowed to disrupt 2 anchorpoints and we are alright with that. -b tells MCScanX that we want both between genomes and within genomes. Try running different options for -s and -m that you think are reasonable and see if the results change much.
Time to visualize the results. We will use provided dot-plot tool written in java. We will have to make three dot plots, two for within each genome and once between.
cd dotPlots
java dot_plotter -g ../moss/moss.gff -s ../moss/moss.colinearity -c Pp_Pp.ctl -o Pp_Pp.png
java dot_plotter -g ../moss/moss.gff -s ../moss/moss.colinearity -c Cp_Pp.ctl -o Cp_Pp.png
java dot_plotter -g ../moss/moss.gff -s ../moss/moss.colinearity -c Cp_Cp.ctl -o Cp_Cp.png
We know that both genome have experienced two rounds of whole-genome duplication, is this evident from their self-syntenic comparisons? Let’s look at the between comparisons, do the ratios between syntenic blocks provide any insights?
More Complex Visualization with Circos
Dot plots are nice because they are intuitive and straightforward enough to write your own plotter in base R for example. But when there are mulitple genomes this can become boring. And what if there are other data you want to display? That is where circos is helpful. It can sometimes be tricky to install, so in lieu of hands-on, I will simply demonstrate the tool.
The example files used for the circos plot based on the MCScanX results are in circos_example. Once you understand some of the basic rules of circos’ control files, it becomes easy to layer on more complexity. The danger with these plots is that is can become too complex to be interpreted by your readers, but that is your call to strike the balance for the audience.
For those interested, we can walk through the install. Circos can be downloaded here. It is completely written in Perl, so does not require compiling, but requires the installation of several Perl modules. I decided to re-install for class and it took me about 10 minutes, but remember my first time took hours…maybe days?