Scaffolding with Hi-C
Hopefully some of the team assemblies are done by this point. Last week, we assembled contigs using various input data and algorithm combinations. Did one strategy or group of strategies appear more beneficial? Ordering those contigs at the correct physical distances on chromosomes though can be difficult, and can greatly affect some analyses of genome architecture or even identifying variants in a population. This week, we will use a scaffolding strategy using high-throughput chromosome conformation capture (Hi-C) data.
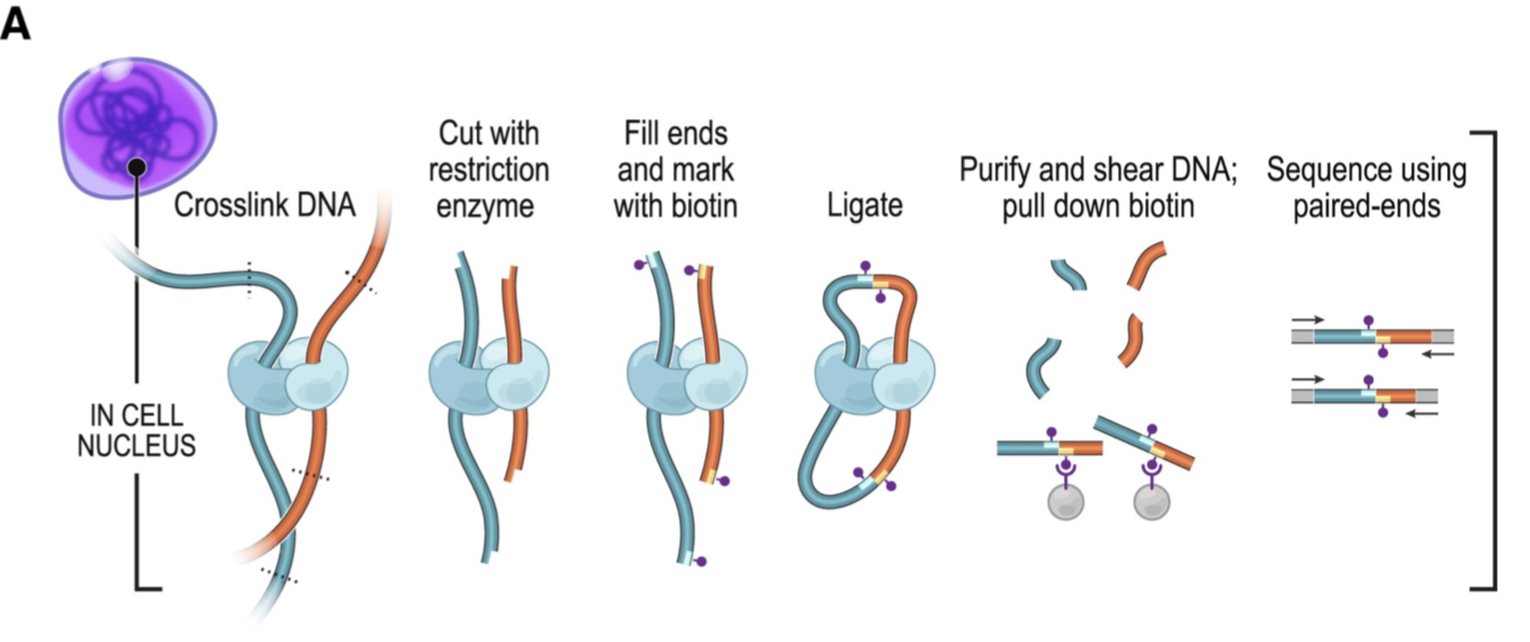
Fig. 1A from Rao et al. 20141- Hi-C library preparation by crosslinking DNA interacting with chromatin and restriction digestion to form a distribution of inserts, some small and some very large.
Although originally intended to investigate DNA-DNA interactions in the cell, such as enhancers and promoter regions coming in contact with each other by forming loops, the range of insert sizes provides the mate information from paired end reads to scaffold entire chromosomes. Although there can be interactions between chromosomes or distal ends of the same chromosome, regions that are physically close together have the most interactions and this is reflected by the frequency that fragments occur in the library. Hi-C libraries can be visualized as 2-d contact maps, where the x- and y-axes are bins of some size and the heatmap intensity is the normalized frequency of a fragment. The scaffolding algorithm finds a path between contigs for scaffolding based on these frequencies. Messy genome assemblies will have contact maps that are very fuzzy while correctly oriented assemblies will generate the most intense signals along the diagonal.
Fig. S6 from Dudchenko et al. 20172- Contact map of human genome. Heat map intensity corresponds to frequency of observed contacts within a bin. Because most interactions happen on the same chromosome, the strongest signals are between adjoining regions. Hi-C can also detect interactions between more distant regions, but the correct path is resolved by the graph weights.
You will continue to run analyses in your /work directory. You should use the contigs from your team’s assemblies last week, and continue to work in teams. Everybody can run the analyses in their own work directory though.
Today’s goals are:
- Generate a file of Hi-C contacts by aligning an Illumina-sequenced Hi-C library to your assembled contigs (Juicer)
- Scaffold those contigs into your final assembly (3-d DNA)
- Inspect the contact maps visually (3-d DNA)
First, we will be doing some copying and uncompressing on the cluster that takes a little more bandwith than we should be using on the head node. Let’s get a processor on the compute node
srun --ntasks=1 --cpus-per-task=1 --mem-per-cpu=4gb --partition=common --account=bio790s-01-f21 --pty bash -i
If you now check your running jobs with squeue -u YOUR_NETID, you should now see an interactive session that says “bash”
We are using tools from the Liberman-Aiden lab and largely following their software tutorials on juicer and 3d-dna. To configure the software requires some careful specification of directories and input files. Much of this has been pre-configured in
/hpc/group/bio790s-01-f21/evolutionaryGenomics/genomeAssembly2
You will want to copy this whole directory into your work directory, but need to pay close attention to edit files when preparing for your own contigs specifying the new work folder location.
Getting Hi-C Contacts from Short-Read Alignment
The first thing you need to do is copy your contigs into the genomeAssembly2/juicedir/references folder. You will then index the genome with BWA. Delete the old fasta and index files if using your own. You will need to edit the submission script for BWA appropriately too. Here are some mock instructions:
cd YOUR_WORK_DIR/evolutionaryGenomics
cp -r /hpc/group/bio790s-01-f21/evolutionaryGenomics/genomeAssembly2 .
cd genomeAssembly2/juicedir/references
cp /YOUR_WORK_DIR/evolutionaryGenomics/genomeAssembly1/PATH_TO_CONTIGS/YOUR_CONTIGS.fasta .
emacs getBWAindex.sh
#sbatch getBWAindex.sh
Genome indexing should be fast, but we will do something else while this happens. Go into the restriction_sites folder, where we will generate a list of expected fragment lengths from the digestion with MboI and a list of contig lengths. Edit the submission script as need be. We use a simple python script provided with juicer to get the restriction site lists. We then use an awk one-liner to get the contig names and lengths, which are the first and last elements per-line resulting from the thrip_MboI.txt file.
cd ../restriction_sites
emacs getRestrictionSites.sh
#sbatch getRestrictionSites.sh
We would normally like to use symbolic links to access fastq files, but this seemed to cause some problems for juicer. Instead, we will all have copies of the fastq files in our work directories. Furthermore they need to be decompressed and renamed as *_R1.fastq and *_R2.fastq
cd ../../HiC-scaffolding/fastq
gunzip SRR11601864_R1.fq.gz
gunzip SRR11601864_R2.fq.gz
mv SRR11601864_R1.fq SRR11601864_R1.fastq
mv SRR11601864_R2.fq SRR11601864_R2.fastq
You should now be ready to run juicer. We will go back to the HiC-scaffolding directory where we will run the juicer script. Be careful to edit it correctly. You will need to provide the correct path of the juicedir -D, the reference fasta -z, the chrom.sizes file -p, and the restriction site fragment size file -y. Provide the correct path to the juicer.sh script in your juicedir/scripts folder too.
cd ../
emacs runJuicer.sh
#sbatch runJuicer.sh
Now we wait a while, BWA has to align the Hi-C reads to the contigs before we can proceed. The runJuicer.sh script actually farms out the jobs on the cluster. These types of things always make me nervous as they take away your control over scheduling directives. This should be ok, but it does not hurt to check with your cluster contacts before running these things for a serious project that mis-allocates a lot of resources.
Scaffolding the Assembly
Juicer should have produced a file called merged_nodups.txt - this is the list of contacts used for building the graph used in scaffolding. The scaffolder takes the original fasta file of contigs and used the contacts to order them. We pass an additional option, -m diploid here since our thrip library is made from a whole pulverized brood. In this case, the whole-genome aligner, lastz, will be used to align and collapse scaffolds that might otherwise be treated differently due to heterozygosity. Edit the script and get 3d-DNA started. There are a lot of options available with the scaffolding algorithm too, have a look at some and see if you want to change some:
/hpc/group/bio790s-01-f21/evolutionaryGenomics/programs/3d-dna/run-asm-pipeline.sh --help
For example, what happens if you increase the mapping quality threshold? Maybe make a few changes that you think will help prevent misjoins.
cd YOUR_WORK_DIR/evolutionaryGenomics/genomeAssembly2/HiC-scaffolding
emacs run3dDNA.sh
#sbatch run3dDNA.sh
This should produce three important files:
- PREFIX.final.fasta
- PREFIX.final.hic
- PREFIX.final.assembly
The fasta file is of course your possibly near-chromosome-level scaffolded genome. The hic and assembly files can be used as input to the visualization software, where it is possible to further annotate the assembly and correct putative errors. Evaluate the basic assembly stats of your PREFIX.final.fasta with quast, how is the N50 looking now?
Visualization
We will do this activity together in real time if allowed. These visualization steps will happen on your own computer rather than the cluster. Be warned that the application has to store a whole genome and its metadata to memory, so the application may crash on some of your laptops.
First, download Juicebox
If we do not make it to the real-time activity, there are many resources available from the Aiden lab to help guide you:
- Introduction to contact maps video
- Step-by-step Juicebox tutorial
- Helpful notes
- Real-time misjoin correction
Try to compare contact maps with others that had a different assembly strategy for generating contigs, how do they look?
References
-
Rao SSP, Huntley MH, Durand NC, Stamenova EK, Bochkov ID, Robinson JT, Sanborn AL, Machol I, Omer AD, Lander ES, Lieberman-Aiden E. 2014. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell. 159:1665-1680. ↩
-
Dudchenko O, Batra SS, Omer AD, Nyquist SK, Hoeger M, Durand NC, Shamim MS, Machol I, Lander ES, Aiden AP, Lieberman-Aiden E. 2017. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science 356:92-95. ↩