ML with IQTREE
Our first day has 2 goals
- Develop basic UNIX skills
- Analyze some sequence data with IQTREE
Remember, struggle and frustration is normal when learning new skills. Please take time to help your neighbors.
If you are not using one of the class computers, there may be some necessary downloads for your own computers in order to participate.
Windows
Mac
- BBEdit
- I recommend downloading the command line tools from the developer. You will need to open a terminal window and type
xcode-select --install. Ask for help if you have not opened a terminal window before. - Although Macs come with Python v2.7, Python 2 is no longer supported. Current and future applications are moving to Python 3 and here is a decent guide for not messing up your system
- R
- IQTREE
- ASTRAL - used tomorrow
- BPP - used tomorrow
Linux
More downloads may be necessary for day 3.
Getting started with the terminal
Can you find “terminal” on the bar with some favorite programs? Go ahead and open it. If this is your first time working in a terminal or command-line environment, it might feel like a very strange way to interact with a computer. However, most phylogenetic software is executed from here. Working in the terminal can be frustrating at first, but with time, it can become very efficient. Let’s practice some basic commands.
Type the following command and then hit enter:
ls
What happened?! You just listed (ls) the directory contents. You will see some different files and folders. One of them are named PhylogenomicsWorkshop.
Let’s go into that folder and have look:
cd PhylogenomicsWorkshop
ls
We used the change directory command (cd) and then list the directory contents. There are currently two folders here:
- workshopPrograms - the programs we will use for analysis have been pre-installed
- workshopData - some data from baobabs we will analyze
A new directory is needed where we can perform our analyses to keep everything tidy.
mkdir analysis cd analysis mkdir ML cd ML lsYou created a new directory named “analysis” with the make directory (mkdir) command. You then change directory into
analysis, again make directoryML, then change directory intoML. We finally list the contents ofMLand see there is nothing.
We can make a file in this folder though with a command-line-based text editor. Try this!
nano bonvoyage.txt
A new screen should open up! You can type something here:
I am a computational biologist
To save the file is a little tricky. You will need to use some keyboard shortcuts:
CTRL + O
ENTER
CTRL + X
That should save the file and close the window. Depending on your keyboard, the CTRL key might actually by ALT. You might have to try a few combinations to get it right.
ls
pwd
We now list the directory contents again and you should see the newly created file. There is also the present working directory command that we use to check where we are at on the computer. As we start to use some programs and analyses, this will be helpful for not getting confused.
Getting started with IQTREE
Let us have a look at the IQTREE program we will use today.
cd ~/PhylogenomicsWorkshop/workshopPrograms/iqtree-2.1.2-Linux/bin
We now change directory into a folder where the IQTREE program exists on the computer. The line might need editing depending on your exact version of IQTREE and where it has been installed. We are going to run the program without any data, just to check that it works.
./iqtree2
Since we did not give it any data, it should show you a help screen with some options. If you got some kind of error, now is a good time to stop and ask for help.
Some example data has been prepared for you. This is some target-enrichment (several hundred nuclear gene sequences) data of baobabs from Karimi et al. (2020)1. People love baobabs and Madagascar is famous for their radiation. However, much of their evolutionary history remains complex. Figure 2 from the original authors is below. Two things to consider is the that biogeographic history of continental African, Australian, and the Malagasy radiation of Adansonia remains unclear. The section Longitubae also appears not monophyletic despite being morphologically similar. The original paper itself gets into complexities beyond this course, but we can still use the data to get used to some phylogenomic analyses.
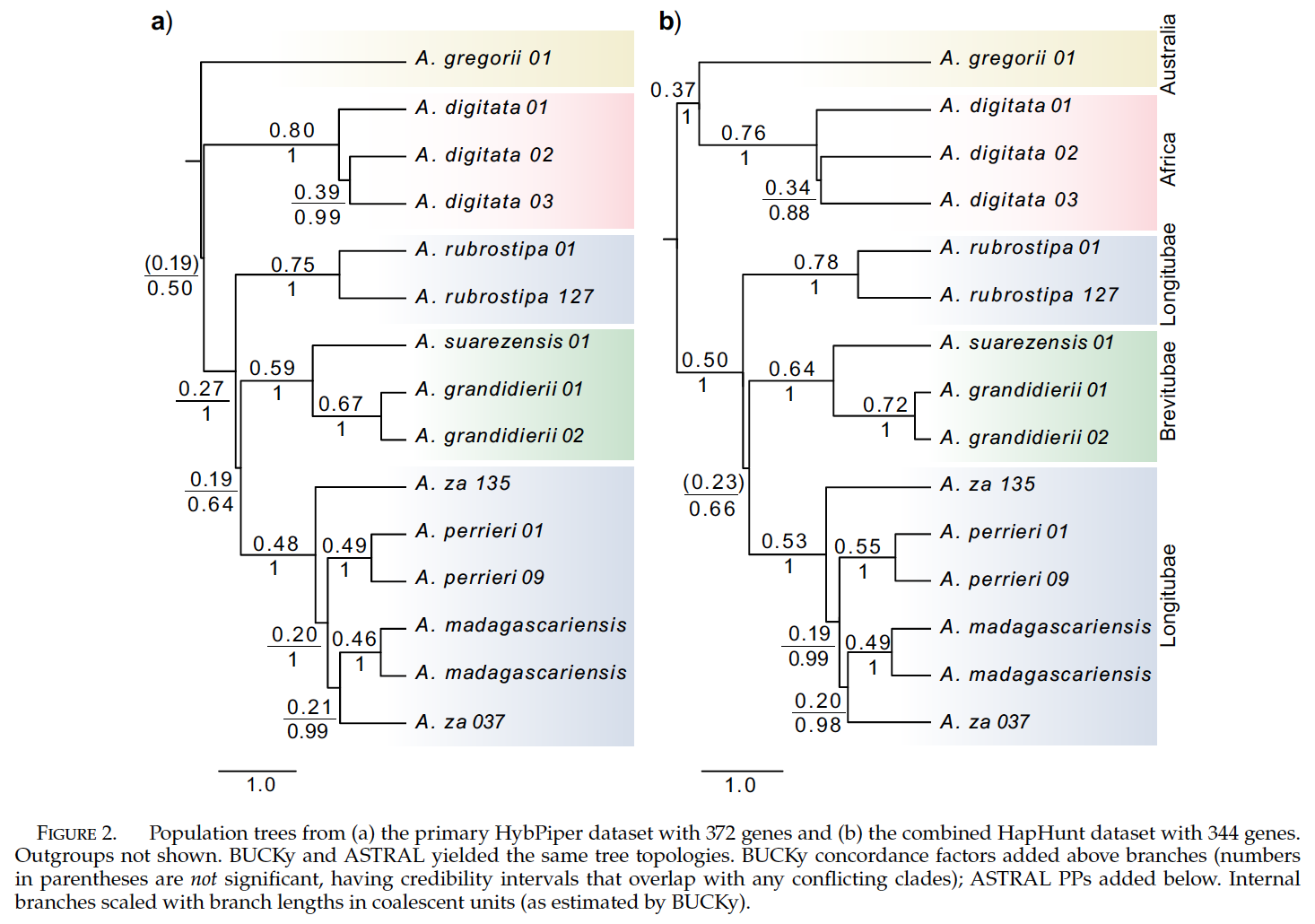
Here is the data
cd ../../../workshopData/ML
ls
I have prepared 10 different subsets for the 10 different Linux stations. This will allow us to compare some results of analyses at the end of class. Please change into your respective directory. If I am at Linux station 1
cd 1
ls
cd oneGene
ls
We will ignore the hundredGenes folder for now. There are three files in the current folder
- YOUR_GENE.fasta - YOUR_GENE can be anything. But, this is a file of aligned protein-coding nuclear sequence from the individuals show in Fig. 2 from Karimi et al. (2020) plus their outgroup Smic.
- partitions.12and3.nexus - This is a NEXUS file used by IQ tree for reading the partition information from you. This file treats first and second codon positions as 1 partition and third codon positions as the 2 partition.
- partitions.1and2and3.nexus - This nexus file assigns first, second, and third codon positions their own partition.
pwd
Can you copy the present working directory? It will help with the next command as we run IQTREE.
TIP! If you are getting tired of typing these very long paths, try hitting the tab (–>) keys as you type and see what happens.
cd ~/PhylogenomicsWorkshop/analysis/ML
mkdir oneGene
cd oneGene
~/PhylogenomicsWorkshop/workshopPrograms/iqtree-2.1.2-Linux/bin/iqtree2 -s ~/PhylogenomicsWorkshop/workshopData/ML/1/oneGene/YOUR_GENE.fasta -B 1000 -m MFP -pre oneGene-unpartitioned
A lot of stuff should start printing to the screens and the analysis will go quickly for a single gene with only a few tips. When the analysis is done, you will see IQTREE created many files. Let’s look at some of the information.
less oneGene-unpartitioned.iqtree
less is a way to read some of the contents of a plain-text file (no special formatting for a program like Microsoft Word). If you scroll down, you will see some information about how many sites were in the alignment and other summary statisitics. You will also see that IQTREE tested 248 models for you! Which one was best and based on which criterion we discussed? Do you see some other features of substitution models that we discussed below?
Continue scrolling down, you will find the maximum log-likelihood. You will also find the Total Tree Length (TL). These are important numbers that we are interested in. Start recording them for your team on this google sheet.
You will also see a text representation of your tree. With time you will understand this format easily and see the rooting, but it can be hard to interpret an unrooted tree sometimes. For now, we will use another program to visualize the tree
java -jar PhylogenomicsWorkshop/workshopPrograms/Figtree/lib/figtree.jar
You just opened the figtree program, which is excellent for visualizing trees and making figures. We will work more with this on Wednesday, but you will notice this a more standard point-and-click program. See if you can open the file oneGene-unpartitioned.treefile with figtree and reroot it with the clade of outgroups (Smi, Pcr,Bce). Take some time to explore options but ask for help if needed. Are Malagasy baobabs monophyletic? Which region is sister to MDG - continental Africa (A. digitata) or Australia (A. gregorii)?
Record the topological result in the google sheet. We are available to help if there are questions about interpreting the result.
Now it is time to repeat the analysis for our partitioned data! Remember to analyses the data for your correct group (i.e. 1, 2, 3, …, 10)
~/PhylogenomicsWorkshop/workshopPrograms/iqtree-2.1.2-Linux/bin/iqtree2 -s ~/PhylogenomicsWorkshop/workshopData/ML/1/oneGene/YOUR_GENE.fasta -B 1000 -m MFP -pre oneGene-12and3 -spp ~/PhylogenomicsWorkshop/workshopData/ML/1/oneGene/partitions.12and3.nexus
Notice there is an extra option added to the command. -spp tells IQtree where to find the file with partition instructions. It also provides some other specifics about the partition model that we will not cover here. You should find very quickly though that more result files have been created. Can you find the same information from before and enter the results into the google sheet?
All that is left is repeating for our model where each codon position is a partition.
~/PhylogenomicsWorkshop/workshopPrograms/iqtree-2.1.2-Linux/bin/iqtree2 -s ~/PhylogenomicsWorkshop/workshopData/ML/1/oneGene/YOUR_GENE.fasta -B 1000 -m MFP -pre oneGene-1and2and3 -spp ~/PhylogenomicsWorkshop/workshopData/ML/1/oneGene/partitions.1and2and3.nexus
If you are this far, then you can analyses 100 genes just as easily as 1!
cd ../
mkdir hundredGenes
cd hundreGenes
ls
pwd
I often use ls and pwd when navigating around to make sure I am in the right place and did not do anything unexpected.
Before we move forward with the analyses, let’s have a look at the data and partition files.
less ~/PhylogenomicsWorkshop/workshopData/ML/1/hundredGenes/concatenated.fasta
Scroll a little bit and you will see this is just a large sequence alignment. Nothing special here. You have seen this with one gene, now it is the same format, only longer.
CTRL + Z
Should let you stop viewing the file. Use less to look at the partition files.
less ~/PhylogenomicsWorkshop/workshopData/ML/1/hundredGenes/partitions.12and3.nexus
CTRL + Z
less ~/PhylogenomicsWorkshop/workshopData/ML/1/hundredGenes/partitions.1and2and3.nexus
CTRL + Z
less ~/PhylogenomicsWorkshop/workshopData/ML/1/hundredGenes/partitions.byGene.nexus
CTRL + Z
The partitions.byGene.nexus file treats each gene separately causing 100 partitions. That’s a lot! We have a strategy for dealing with this for the final analysis. IQTREE allows you to add +MERGE to the model option which will make the software explore ways to reduce the number of partitions.
Here are the final IQTREE commands, but take your time. Look through the results, think about the differences in analyses. Maybe try to type them yourself and not copy and paste to get some practice with IQTREE’s options?
~/PhylogenomicsWorkshop/workshopPrograms/iqtree-2.1.2-Linux/bin/iqtree2 -s ~/PhylogenomicsWorkshop/workshopData/ML/1/hundredGenes/concatenated.fasta -B 1000 -m MFP -pre hundredGenes-unpartitioned
~/PhylogenomicsWorkshop/workshopPrograms/iqtree-2.1.2-Linux/bin/iqtree2 -s ~/PhylogenomicsWorkshop/workshopData/ML/1/hundredGenes/concatenated.fasta -B 1000 -m MFP -pre hundredGenes-12and3 -spp ~/PhylogenomicsWorkshop/workshopData/ML/1/hundredGenes/partitions.12and3.nexus
~/PhylogenomicsWorkshop/workshopPrograms/iqtree-2.1.2-Linux/bin/iqtree2 -s ~/PhylogenomicsWorkshop/workshopData/ML/1/hundredGenes/concatenated.fasta -B 1000 -m MFP -pre hundredGenes-1and2and3 -spp ~/PhylogenomicsWorkshop/workshopData/ML/1/hundredGenes/partitions.1and2and3.nexus
~/PhylogenomicsWorkshop/workshopPrograms/iqtree-2.1.2-Linux/bin/iqtree2 -s ~/PhylogenomicsWorkshop/workshopData/ML/1/hundredGenes/concatenated.fasta -B 1000 -m MFP -pre hundredGenes-byGene -spp ~/PhylogenomicsWorkshop/workshopData/ML/1/hundredGenes/partitions.byGene.nexus
~/PhylogenomicsWorkshop/workshopPrograms/iqtree-2.1.2-Linux/bin/iqtree2 -s ~/PhylogenomicsWorkshop/workshopData/ML/1/hundredGenes/concatenated.fasta -B 1000 -m MFP+MERGE -pre hundredGenes-byGene -spp ~/PhylogenomicsWorkshop/workshopData/ML/1/hundredGenes/partitions.byGene.nexus
###End You should now feel more comfortable navigating the terminal and running some software from it. Many topics from the lecture should have been identifiable in the output from IQTREE. We have been very short on the details to keep material practical, but the more often you analyze data, you will become more comfortable with both the theory and application.
-
Karimi N, et al. 2020. Reticulate evolution helps explain apparent homoplasy in floral biology and pollination in baobabs (Adansonia; Bombacoideae; Malvaceae). Systemaic Biology. 69:462-478. ↩